Als Erweiterung des vorangegangenen Projektes habe ich im Zuge meiner Dissertation mit einer erhöhten Anzahl an Drachenkopf Arten und Akzessionen Daten zusätzlicher genetischer Marker erhoben und verglichen. Eines der Ergebnisse ist ein diagnostischer Primer, der den nahezu artspezifischen Nachweis des Moldawischen Drachenkopfes zulässt. Des weiteren konnten deutliche Unterschiede in der genetischen Vielfalt zwischen den untersuchten Moldawischen Drachenköpfen und z.B. den nordischen Drachenköpfe festgestellt werden.
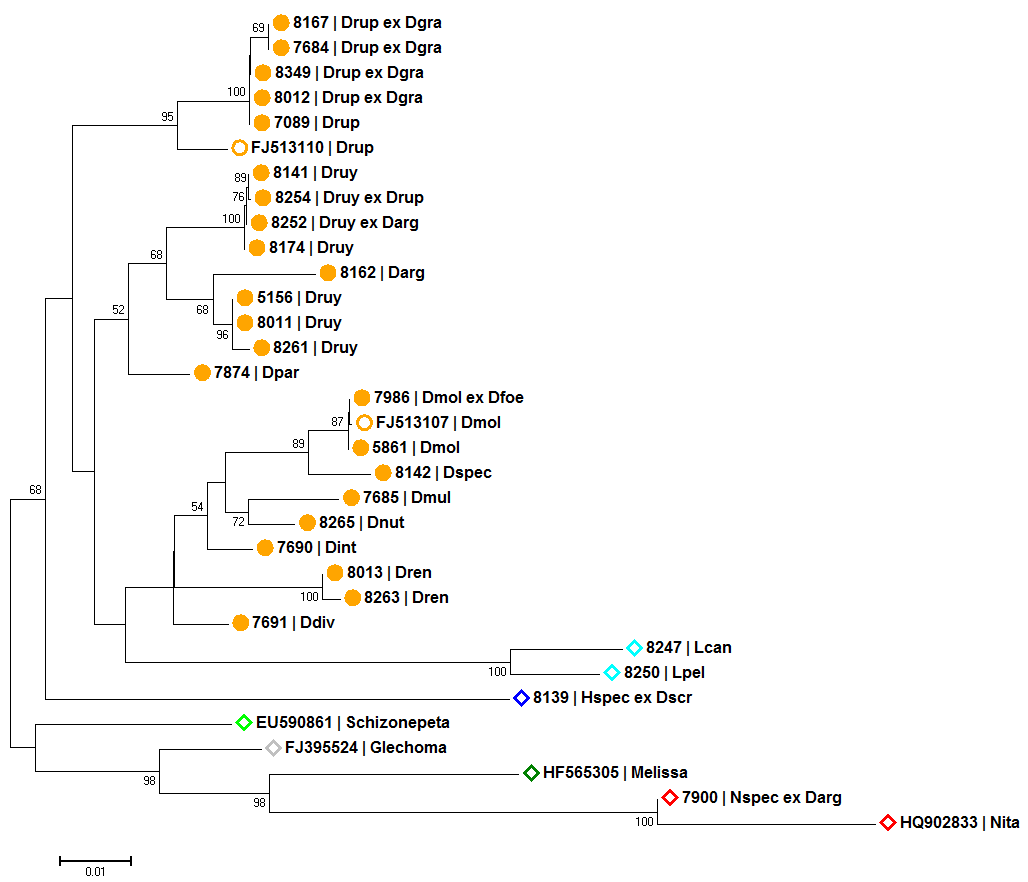
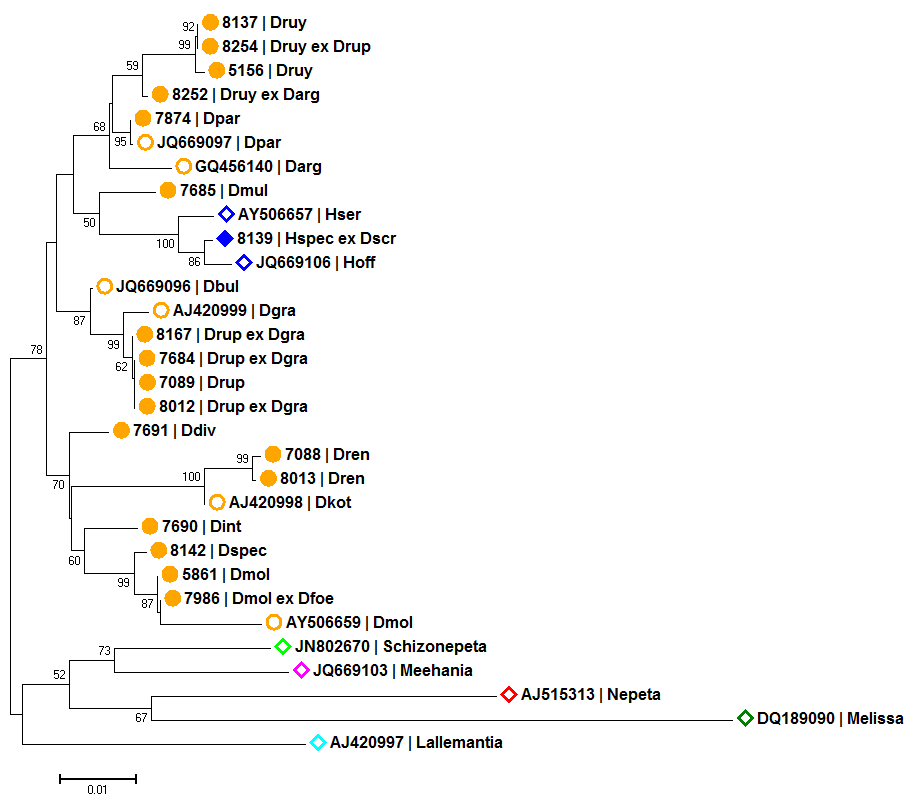
Genetische Distanzen in Form von phylogenetischen Bäumen
Genetische Unterschiede zwischen homologen DNA Bereichen kann man in Zahlen ausdrücken, wie z.B. der proportionalen Distanz (p-distance). Im Vergleich vieler verschiedener Sequenzen dieser homologen Bereiche entsteht eine Distanz Matrix die mit Hilfe von Programmen, wie z.B. MEGA, visuell als phylogenetischer Baum dargestellt werden kann. Dabei finden sogenannte clustering algorithmen, wie z.B. Neighbour-Joining, Anwendung.
Als Ergebnis der Analysen erwiesen sich zwei Marker Bereiche als ausreichend variabel, um die von uns untersuchten Arten genetisch zu unterscheiden:
- Neighbour-Joining Baum basierend auf dem Plastid Marker psbA-trnH und dem Kern Marker Internal Transcribed Spacer (ITS)
{kind=link}
{kind=link}
Variabilität verschiedener DNA Barcoding Marker im Vergleich
Mit Hilfe des Programms TaxonGap kann man zum einen genetische Ähnlichkeit (similarity) eines Marker Bereichs im Vergleich verschiedener Arten darstellen und zum anderen einen Vergleich zwischen verschiedenen Marker Bereichen erstellen.
- Vergleich des Internal Transcribed Spacer (ITS) 1, der 5.8 S rDNA sowie des ITS 2 (Kerngenom)
- Vergleich eines Bereichs des Ribulose-bisphosphate Carboxylase Gens (rbcL), des maturase K (matK) Gens sowie des intergenic Spacers psbA-trnH (Plastiden Genom)
{kind=link}
{kind=link}
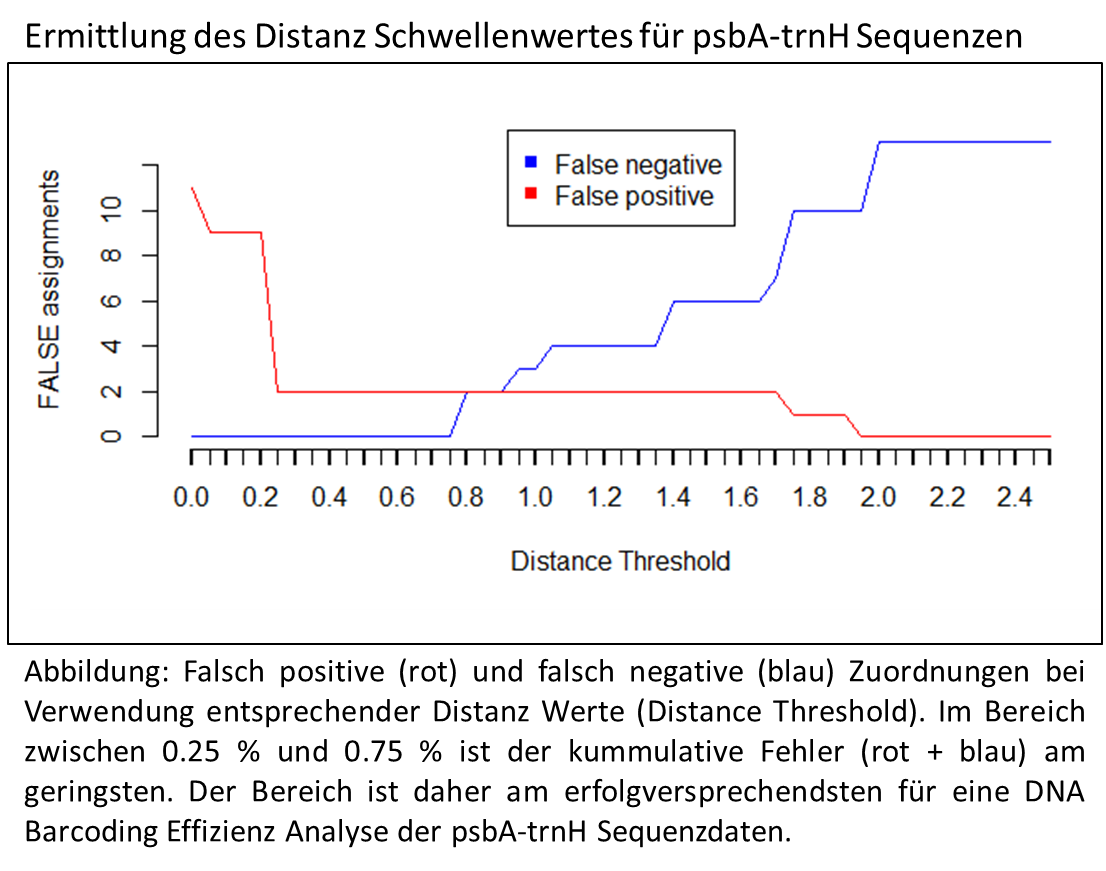
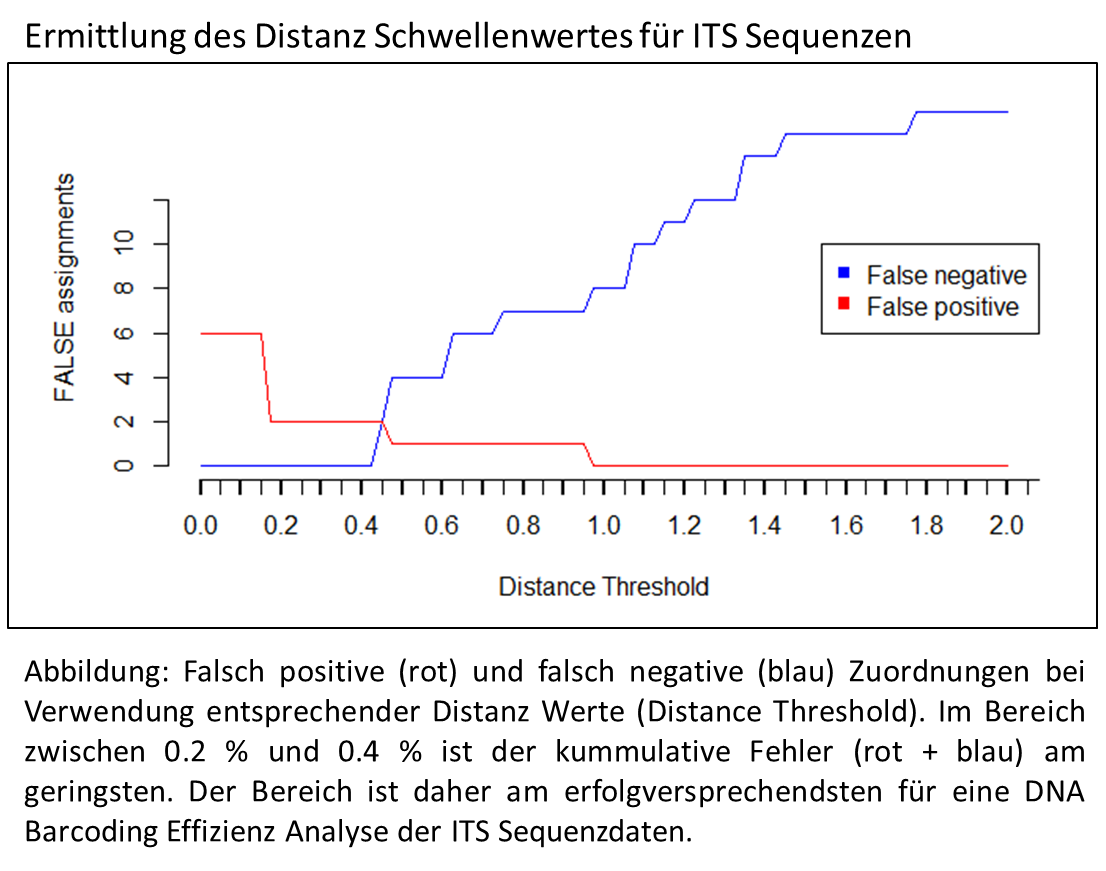
Nützlichkeit genetischer Marker beim DNA Barcoding
Das Ziel des DNA Barcoding ist es, einen oder mehrere genetische Abschnitte zu finden, deren Sequenz Information eine eindeutige Art Zuweisung erlaubt. Mit Hilfe von Algorithmen lassen sich entsprechende Sequenz Sammlungen prüfen. Jede Sequenz wird dabei einmal als unbekannte Probe verwendet und mit den anderen verglichen. Je nach Algorithmus wird die taxonomische Zugehörigkeit, z.B. die der ähnlichsten Sequenz, der unbekannten Probe zugeordnet. Da die taxonomische Zugehörigkeit vorher schon bekannt ist kann man nach Prüfung aller Sequenzen sagen, wie viele der Sequenzen zugeordnet werden konnten und bei wie vielen dies erfolglos (falsche Zuordnung) verlief.
Da die vorhergehende Analyse der genetischen Distanz und Variabilität gezeigt hatte, dass psbA-trnH und ITS die vielversprechendsten der 4 getesteten Marker sind, wurde auch nur mit diesen eine entsprechende Analyse durchgeführt.
Manche Algorithmen erlauben die Einstellung eines Distanz Schwellenwertes. Dies bedeutet, dass für die taxonomische Zuordnung nur die Sequenzen berücksichtigt werden, die innerhalb einer bestimmten genetischen Distanz liegen.
{kind=link}
{kind=link}